
Statistical genomics require rapid rehashing of a central, consistent dataset against an ever-evolving set of variables to strike correlations. This central consistent dataset is often fairly small and serves as a reference to seek variability between genomes. Every sequenced genome is the product of an extensive computational process that transforms hundreds of gigabytes of raw sequencer output into aligned genomes, and the sequencing industry has been demanding increasingly larger compute and storage resources to keep up.
Data parallelism and statistical methods lie at the core of all DNA sequencing workloads because the physical process of decoding DNA is error-prone. These errors and uncertainties of errors are effectively mitigated by sequencing the same pieces of DNA many times over and calculating what is referred to as a “quality score”. This is an indication of how likely any given data point may be an error. At a very high level, the general process working with sequenced DNA can be broken down into three steps each with their own storage and compute requirements.
A standard genomics pipeline involves converting BCL files, written from sequencer to a Windows client. This process coverts raw data into millions of short reads or base calls. The data often lands on Network Attached Storage with limited, if any, benefit to running this on a parallel file system. Typical runs for a NGS system are in the 10’s to 100’s of MB/s and can last for a day or two.
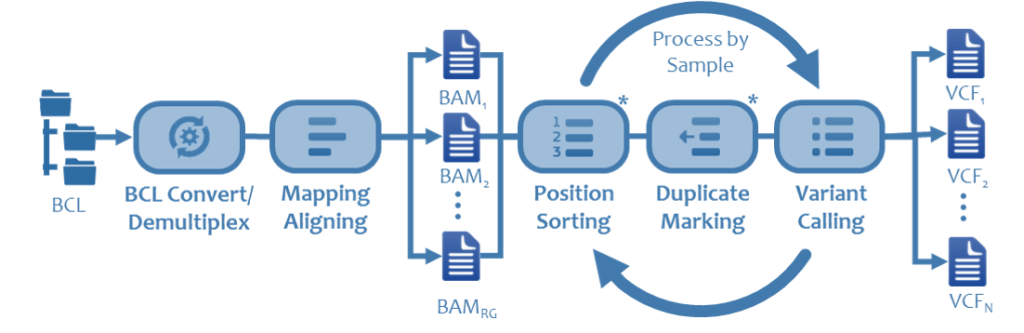
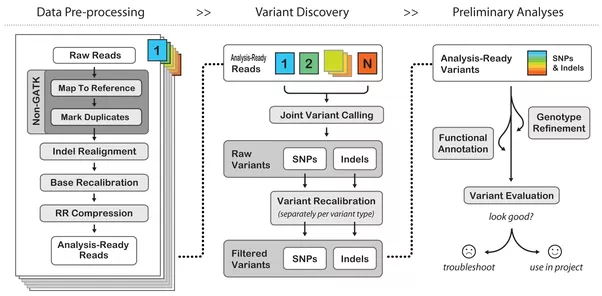
Once the base calls are generated, they are mapped to a reference genome. This is referred to as the alignment process and determines how the base calls fit together. Variants, or reads that do not match are identified in a process called variant calling and saved in a separate dataset. This step requires significant capacity both in-flight and post processing. The amount of temporary capacity ranges between 8 and 10 times its input, and output data is typically 2 -3 times the size of the original dataset. This step scales linearly with cores and while the dataset is robust, does not take advantage of parallel IO.
Once the variant calling process is complete, the real science begins, taking the called variants and correlating those differences with scientific theory. The process often involves cross-referencing the called variants with other known variants or other analysis. Depending on the code and libraries used, the process takes significant advantage of cluster resources and high performance parallel file systems.
